# A tibble: 344 × 8
species island bill_len bill_dep flipper_len body_mass sex year
<fct> <fct> <dbl> <dbl> <int> <int> <fct> <int>
1 Adelie Torgersen 39.1 18.7 181 3750 male 2007
2 Adelie Torgersen 39.5 17.4 186 3800 female 2007
3 Adelie Torgersen 40.3 18 195 3250 female 2007
4 Adelie Torgersen NA NA NA NA <NA> 2007
5 Adelie Torgersen 36.7 19.3 193 3450 female 2007
6 Adelie Torgersen 39.3 20.6 190 3650 male 2007
7 Adelie Torgersen 38.9 17.8 181 3625 female 2007
8 Adelie Torgersen 39.2 19.6 195 4675 male 2007
9 Adelie Torgersen 34.1 18.1 193 3475 <NA> 2007
10 Adelie Torgersen 42 20.2 190 4250 <NA> 2007
# ℹ 334 more rowsElements of Data Science
SDS 322E
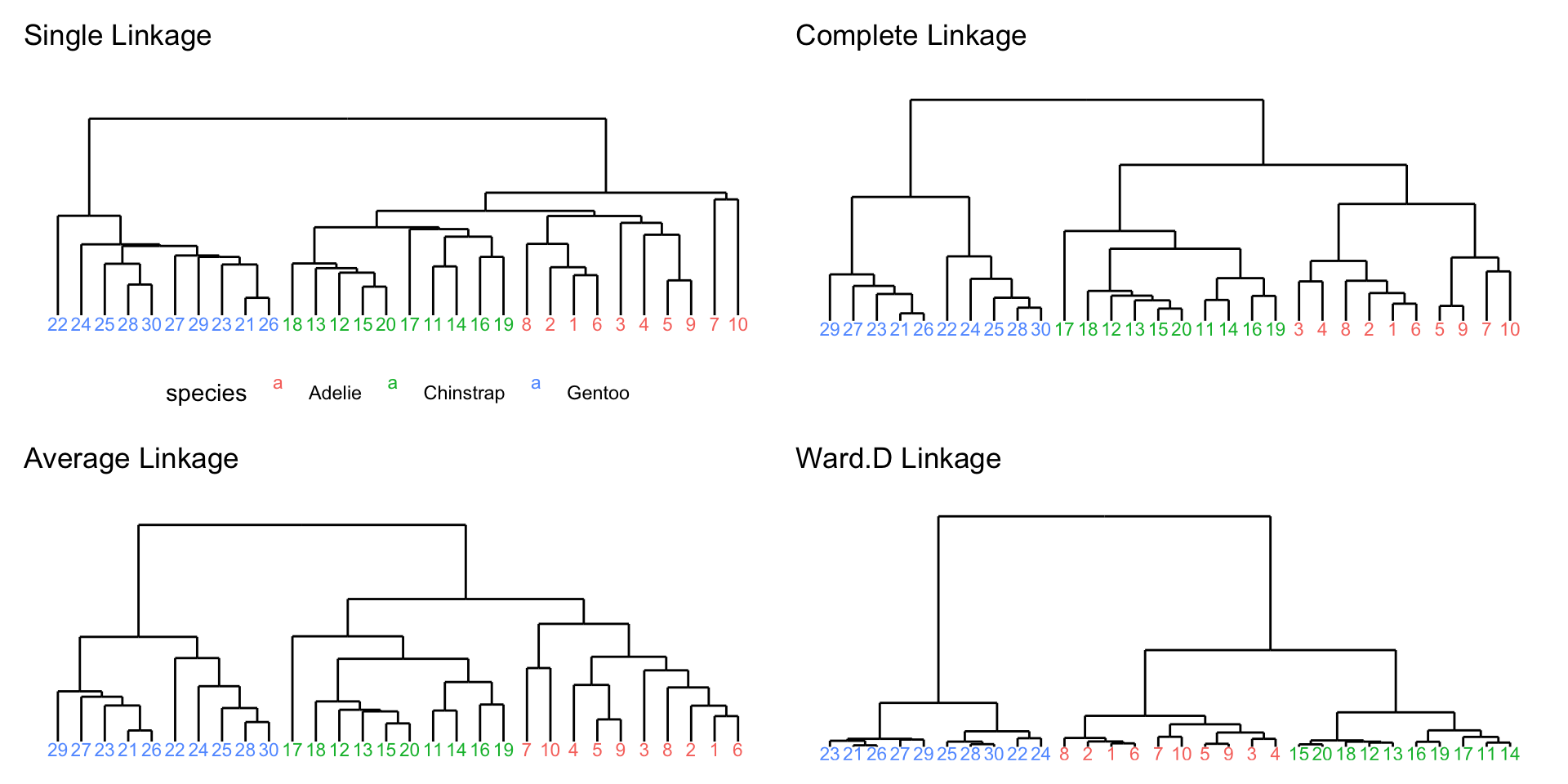
Visualize hierarchical clustering results
Visualize hierarchical clustering results
Code
hclust_data1 <- ggdendro::dendro_data(hclust_results1)
hclust_segments1 <- as_tibble(hclust_data1$segments)
hclust_labels1 <- as_tibble(hclust_data1$labels) |>
mutate(species = penguins_small$species[as.integer(label)])
p1 <- ggplot() +
geom_segment(data = hclust_segments1,
aes(x = x, y = y, xend = xend, yend = yend)) +
geom_text(data = hclust_labels1,
aes(x = x, y = y - 0.02, label = label,
color = species), vjust = 1, size = 3) +
labs(title = "Single Linkage") +
theme_minimal() +
theme(axis.title = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
panel.grid = element_blank(),
legend.position = "bottom") +
ylim(-0.1, 2.5)
p1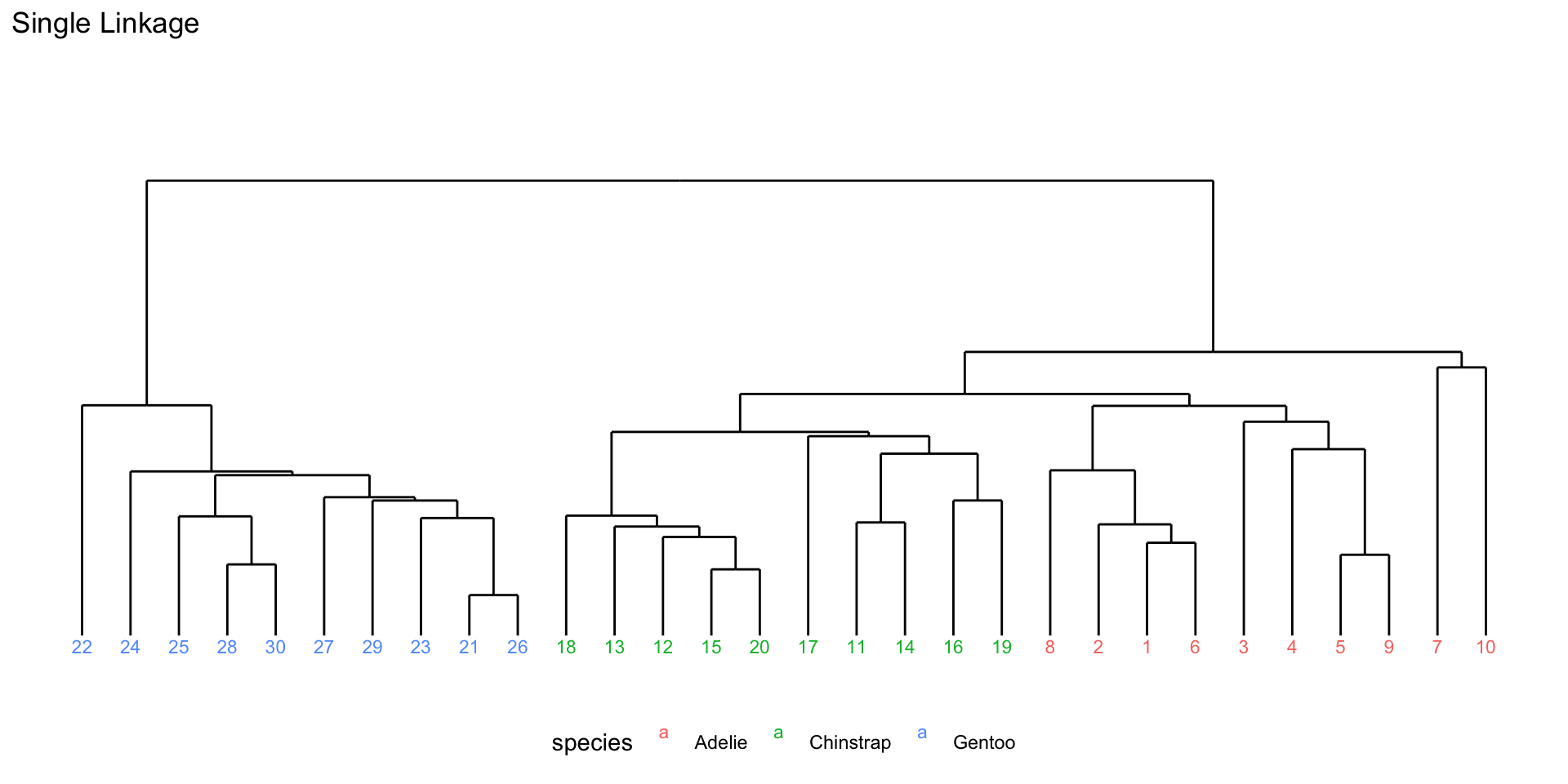
Different linkage for penguins data
Full penguins data
Code
hclust_data <- ggdendro::dendro_data(hclust_results)
hclust_segments <- as_tibble(hclust_data$segments)
hclust_labels <- as_tibble(hclust_data$labels) |>
mutate(species = penguins_clean$species[as.integer(label)])
ggplot() +
geom_segment(data = hclust_segments,
aes(x = x, y = y, xend = xend, yend = yend)) +
geom_text(data = hclust_labels,
aes(x = x, y = y - 0.02, label = label,
color = species), vjust = 1, size = 3) +
labs(title = "Average Linkage") +
theme_minimal() +
theme(axis.title = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
panel.grid = element_blank(),
legend.position = "none") +
ylim(-0.1, 4) 
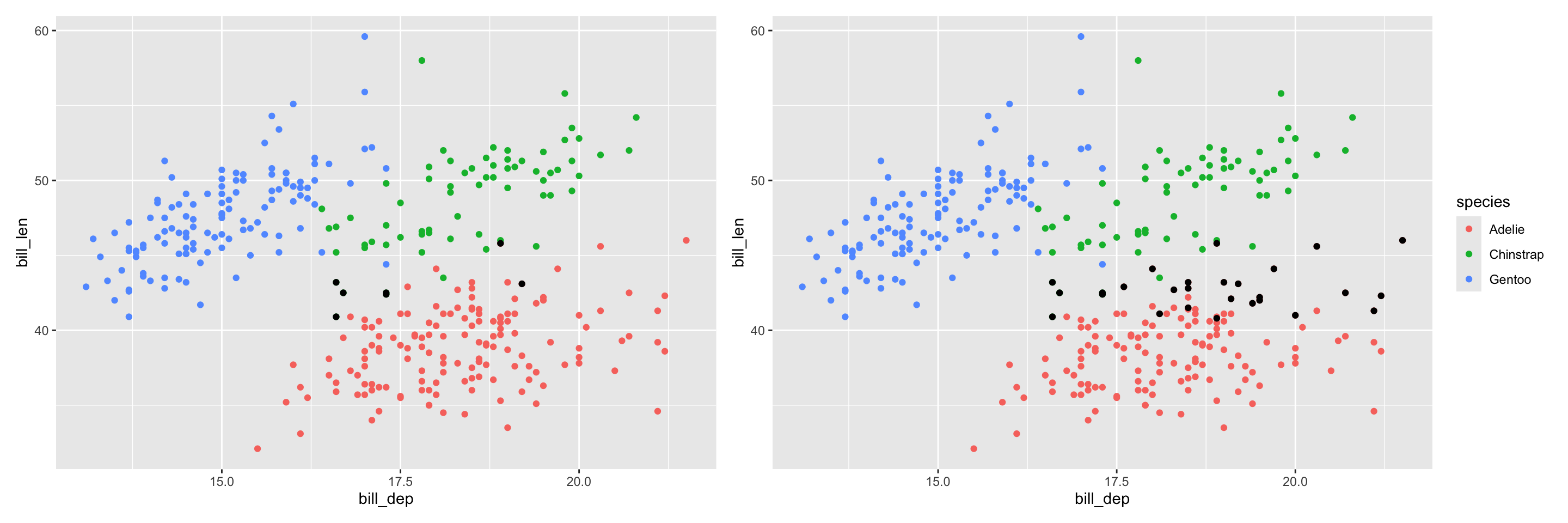
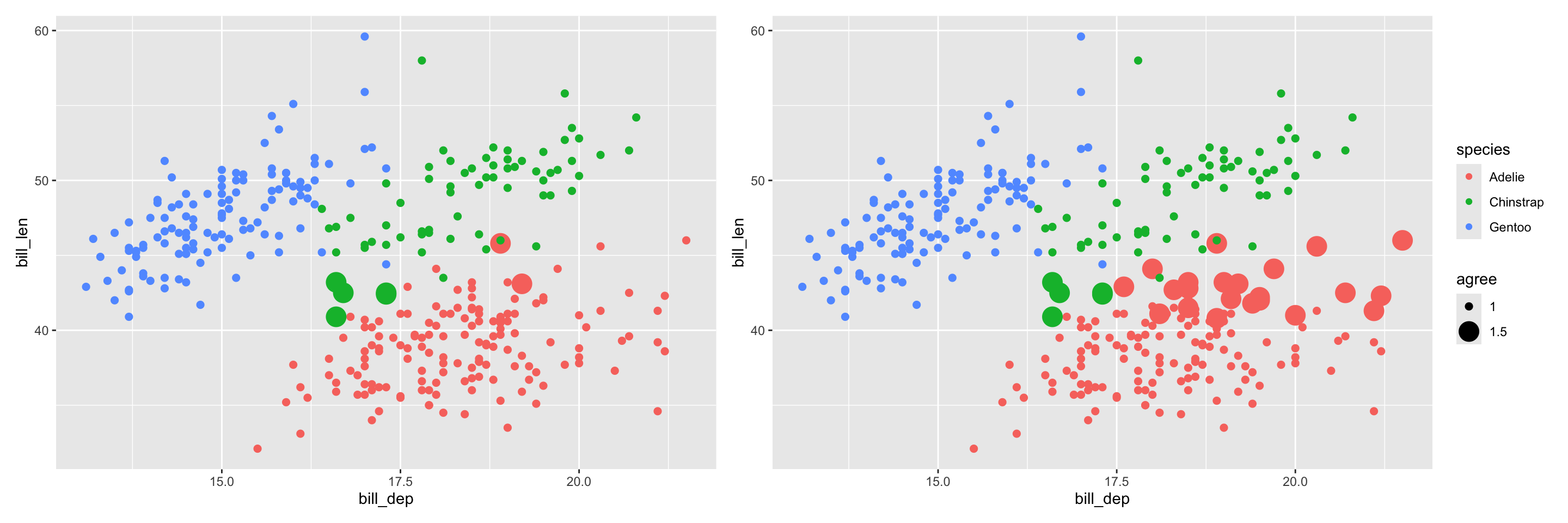
Cut the tree into two clusters
The base R function cutree() can be used to cut the dendrogram into a specified number of clusters.
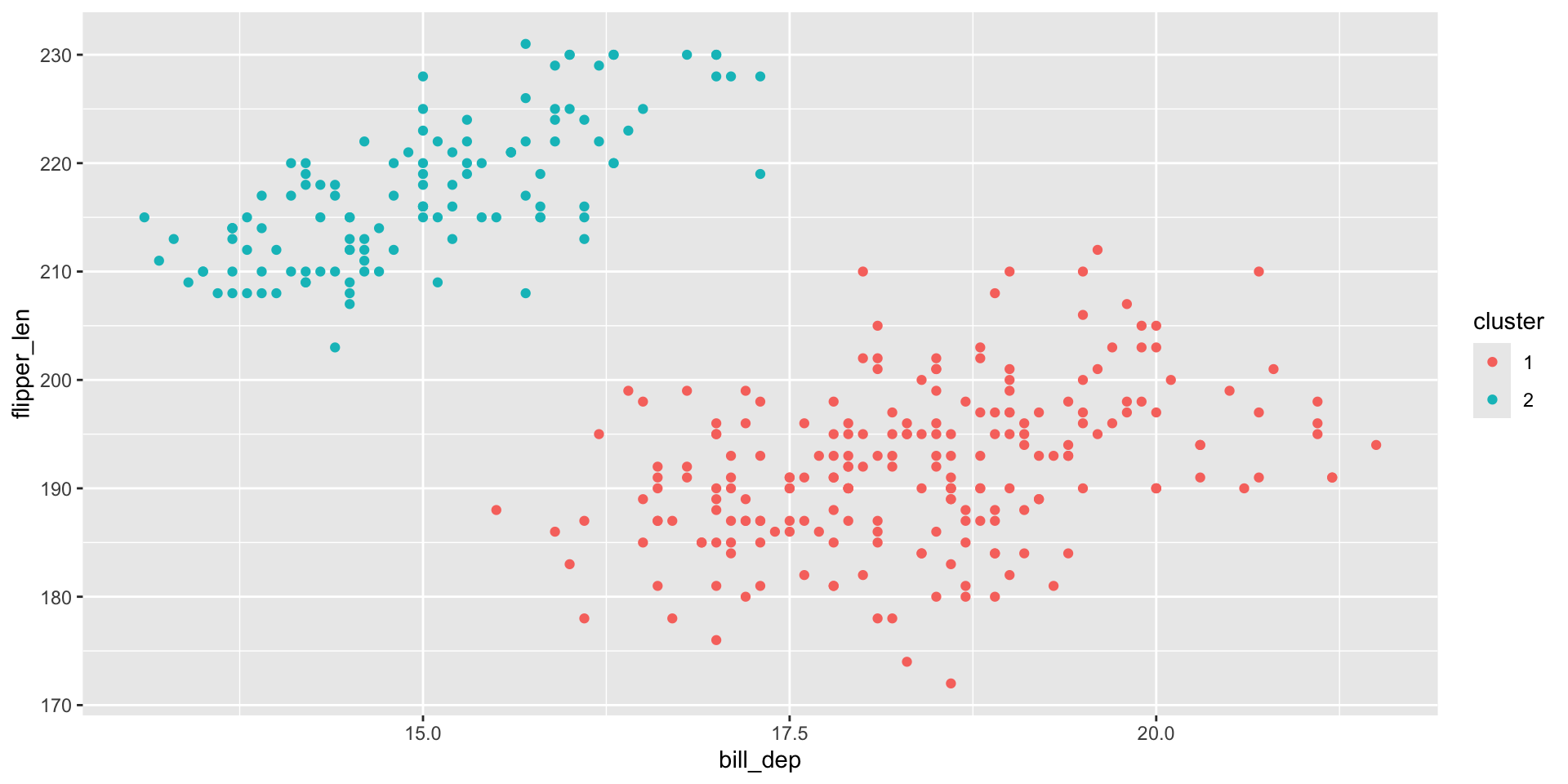
Compare hierarchical clustering with kmeans
Hierarchical clustering: cut into 3 groups
# A tibble: 5 × 3
cluster species n
<fct> <fct> <int>
1 1 Adelie 144
2 1 Chinstrap 5
3 2 Adelie 2
4 2 Chinstrap 63
5 3 Gentoo 119Kmeans clustering: choose 3 centers
# A tibble: 5 × 3
cluster species n
<fct> <fct> <int>
1 1 Adelie 124
2 1 Chinstrap 5
3 2 Adelie 22
4 2 Chinstrap 63
5 3 Gentoo 119